重磅!官媒再发声:我国细胞治疗终于进入临床的大门!
据北京市卫生计划委员会报道,北京市近日会同有关部门,组织干细胞临床研究备案机构,召开了干细胞临床研究监督检查现场会,对控制干细胞临床研究质量和风险有效管控等方面提出具体要求。
近日,多个免疫疗法产品申报临床连同细胞治疗产品技术指导原则发布,被评为2017年中国医药生物技术十大进展。这一指导原则为什么被整个行业翘首以待,又对哪些事情进行了明确?
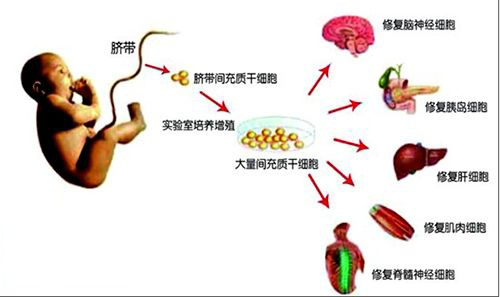
对癌症和遗传病治疗说“yes”
自2016年12月《细胞制品研究与评价技术指导原则》(征求意见稿)发布之后,细胞治疗产品一直在等待通往临床大门的开启,为癌症治疗、遗传病治疗给出“yes”的答案。直到2017年12月18日,国家食药监总局发布了《细胞治疗产品研究与评价技术指导原则(试行)》,被产业界评为“靴子终于落地”。从最终发布的文件名称上看,原来的“细胞制品”改为“细胞治疗产品”,可以看出该指导原则意在推动相关疗法的应用。
“指导原则的发布,让细胞治疗向临床产品的目标更近了一步,”专家说,这意味着,如果申请企业的细胞制品符合要求,经过临床试验证明有效,应该不久就会批准下来,细胞治疗“落地”不会远了。

自2017年8月底,美国FDA批准诺华公司的细胞免疫疗法上市以来,我国产业界对细胞治疗的上市一直充满期待,国家食药监总局发布《细胞治疗产品研究与评价技术指导原则(试行)》,意味着进入临床的大门终于打开。
安全性、有效性和质量可控性水平更高
“原来模棱两可的操作有了明确的标准,”专家说,“之前行业内的标准是行业协会的要求,是一种自发的约束,有的团队比行业标准严格,有的可能不如团队标准。”标准的实施进一步规范了细胞治疗产品的研发,提高其安全性、有效性和质量可控性水平,推动和促进我国细胞治疗领域的健康发展。
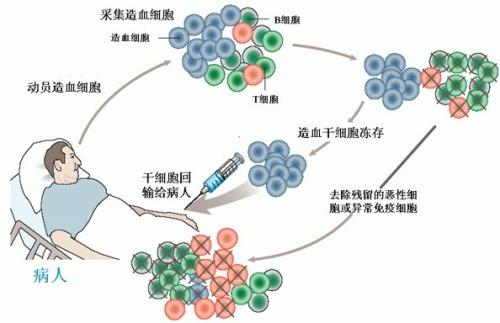
“由于细胞治疗产品的特殊性,此前新药的评价体系很难照搬套用。”专家说,“药片是死的,细胞是活的,比如干细胞,它培养繁殖会产生不同的‘代数’,不同代数细胞干性不同,其治疗的功能性就会有差异。”
从制备到使用的时间间隔上,细胞治疗产品也和药品有着很大区别。“药片的保质期可以是几年,而细胞制品上午制备出来,下午就能给病人回输,但是质量控制的检验结果可能明天才出来,怎么办呢?”

由于与药品存在很大不同,此前的细胞治疗产品中的免疫细胞治疗产品参照过“第三类医疗技术”进行管理,业内也有按照“新药”和“医疗技术”两种不同的管理声音。“指导原则”的发布则明确细胞治疗产品按照“新药”申报管理。“选择这条路更加严格,确保安全性、有效性和质量可控性。”专家说。
而针对细胞治疗产品的个性化、时效性等特点,“指导原则”除了规定严格的生产工艺、操作规范等,还引入预案的解决方式,文件中写到:建议尽量在产品临床应用前完成全部放行检验,当有些放行检验结果可能后置时,应对可能出现的非预期检验结果制订处置方案。
此外,细胞制品本身有着易污染、无法耐受病毒细菌灭活处理的特点,加强过程控制成为生产规范中的重点。在药监局相关解读中强调“全程监控”,细胞治疗产品的每一个生产步骤均应该进行研究与验证,以保证工艺的合理性和稳定性。“同时要制定预案,被污染了或者有病原体接入了的情况,都要有预案应对。”专家说。
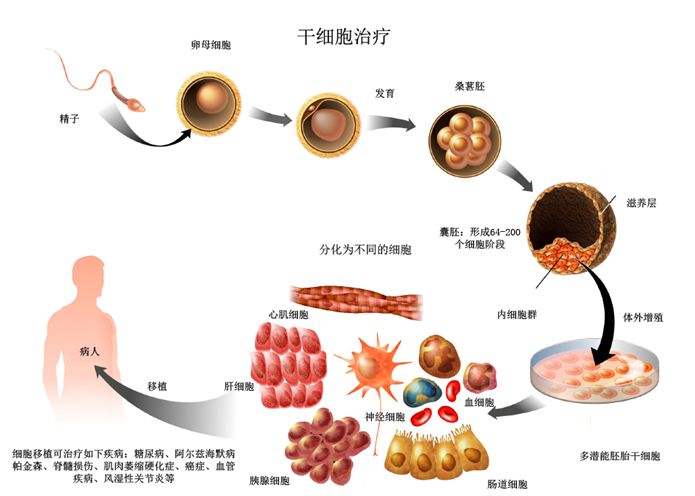
对具体指标进行数字化规范
除了整个过程的控制,细胞制品的质量控制是分“两头”的。专家介绍,“包括原材料和成品之后两方面的质量控制。”
“指导原则”还对之前模棱两可的做法做了明确,例如如何评价细胞治疗产品的致瘤性、致癌性。这是一个预估的命题,因为一些治疗过程中使用的生长因子可能引起宿主细胞或细胞治疗产品本身发生肿瘤,尽管传统的致癌性试验可能不适应于细胞治疗产品,但由于目前尚未达成科学共识,此次指导原则仍沿用传统。
再比如,根据此前的新药审评审批流程,已经在人体上有效果的治疗,有时还需要返回进行动物实验的验证,这对科研团队来说并不合理。此次原则明确了“人体数据在经过科学地评估后,可以提示细胞治疗产品的有效性与安全性,可以保证临床受试者安全性,则可免除不必要的动物试验”。
“未来还会制定更加明确、详细的要求,”专家说,“例如,对活度、标的物等具体指标给出一个数字化的规范。”

